


SARS-1: Evidence of an Artificial Origin
As the world debates the origin of SARS-COV-2, most assume the SARS outbreak of 2003 was a natural event. But revisiting the evidence I found parallels, direct linkages and many unresolved questions.


To understand the origin of SARS-CoV-2 it’s illuminating to revisit the history of the first SARS outbreak and the subsequent investigation of its origin. SARS is assumed to be the result of a natural zoonosis by most scientists - including many who are open to an artificial origin of SARS-CoV-2. But the basis for this assumption may be unsound. There are many parallels between the two viruses both at the molecular level, and also in the epidemiology, pandemic management and origins tracing. Many of the same individuals and institutions play key roles in both. It may be that these outbreaks aren’t independent events, but the result of a long-term research program.
History of the Outbreak
The first known case of SARS is recorded (retrospectively) on 16th November 2002, the day after Hu Jintao assumed the role of general secretary of the CCP, effectively China’ leader. Hu’s predecessor Jiang Jemin had a record as a reformer. Under Jiang China’s market economy had grown rapidly, trade, diplomacy and international investment had blossomed. In contrast Hu's reputation was built on the violent suppression of Tibetan nationalism, placing it under military rule. His ascendancy signaled a slowing of the pace of China's reform and opening up to the world.
The outbreak is believed to have started in Guangdong, although the first case wasn’t diagnosed until February 28th, 2003 in Vietnam by Dr Carlo Urbani, who died of the disease a month later. The Chinese official response was characterized by secrecy and misinformation. Initially the government denied there was an infectious disease involved at all. The number and severity of cases was kept hidden:
“It was difficult to know what was going on in the mainland, especially where a sensitive issue like the emergence of a new disease was concerned. No announcements were made either by Beijing or by local authorities in Guangdong Province. However, the controlled media did, at times, publish articles from which some information could be gleaned. Thus, Guangdong newspapers in early January 2003 published a handful of articles denying the existence of any epidemic but, by doing so, enhanced speculation of a deadly new disease.”
“The Chinese public, and the rest of the world, was kept in ignorance. Under Chinese law, any occurrence of infectious diseases should be classified as a state secret before they are “announced by the Ministry of Health or organs authorized by the Ministry.” That is to say, until the government made the information public, any doctor or journalist who disclosed information on the disease was liable to prosecution for leaking state secrets”
“…a circular appeared in the local media that acknowledged the presence of the disease and listed some preventive measures, including improving ventilation, using vinegar fumes to disinfect the air, and washing hands frequently.”
Email to WHO team leader: “Am wondering if you would have information on the strange contagious disease (similar to pneumonia with invalidating effect on lung) which has already left more than 100 people dead in Guangdong Province, in the space of one week. The outbreak is not allowed to be made known to the public via the media, but people are already aware of it (through hospital workers) and there is a ‘panic’ attitude, currently, where people are emptying pharmaceutical stocks of any medicine they think may protect them.”
Eventually it was officially declared that a chlamydia bacterium was responsible:
“March 19, WHO received a letter from China’s Ministry of Health announcing that chlamydia was found by electron microscopy in five SARS patients. Actually, as early as February 19, the Ministry of Health had said to the WHO: “It is almost ascertained that the causal agent for the atypical pneumonia outbreak in Guangdong is chlamydia.” This was based on the work of Hong Tao, a senior microbiologist at China’s CDC.”
Despite this pronouncement, and before the epidemic had reached Hong Kong, researchers from HKU and an international network setup by WHO, raced to obtain samples and identify a possible viral pathogen. HKU eventually succeeding in isolating the novel coronavirus after experimenting with different cell lines:
“Dr. Chan decided to use a cell line of fetal kidney cells from rhesus monkeys. It was rarely used except to grow hepatitis A virus but it had also proved useful in growing a range of respiratory viruses…Two days later there was a visible reaction as the cells appeared to be dying…there was a virus taking over… “The use of this cell line (FRhk-4) was probably the most important decision in the discovery of the pathogen behind SARS,” Yuen and Peiris wrote.”
“It is not one of the two known human coronaviruses and not even any animal coronavirus,” Professor Peiris said. “We are dealing with a type of virus which we have never come across before”
Later it emerged that the PLA’s Academy of Military Medical Sciences (AMMS) in Beijing had for some time known a novel coronavirus was to blame, but had not disclosed it. The reason? It “would not have been respectful” to contradict the official line:
“Yang Ruifu, a soft-spoken microbiologist and a member of the team at the Academy of Military Medical Sciences (AMMS) that discovered the coronavirus. Promoted by Hong Tao, an esteemed senior microbiologist and member of the Chinese Academy of Engineering, the Chlamydia hypothesis had become so well established that “it would not have been respectful” to challenge it, Yang says. Indeed, others say, the Ministry of Health had effectively banned alternative views.”
“These [AMMS] scientists were the first ever to see the SARS virus,” Klaus Stohr of the WHO said after visiting the academy, “and we had no idea.”
Academy of Military Medical Sciences
The PLA's Academy of Military Medical Sciences (AMMS) is rarely identified on scientific papers authored by its officers, who prefer to use affiliations of related entities that sound less obviously "military" i.e. State Key Laboratory of Pathogen and Biosecurity, or Beijing Institute of Microbiology and Epidemiology ("soft-spoken microbiologist", and also plague expert, Yang Ruifu is now the director of these). AMMS Officers usually use civilian honorifics like Doctor or Professor in preference to military rank. Some, like Wuchun Cao, Changchun Tu and Yusen Zhou (who recently died in mysterious circumstances), are well networked with western scientists, have studied and worked abroad and collaborated widely.
But AMMS has long been identified by the US State Department as likely engaged in covert offensive bioweapons development in contravention of international agreements. Its leaders have been known for dark and disturbing think pieces and speeches. And it has been engaged in a decades long program to collect and study novel pathogens.
US bioweapons compliance reports identify by name a second Chinese state entity - Lanzhou Institute of Biological Products. Its sister company Wuhan Institute of Biological Products (WIBP) is situated next door to WIV's BSL-4 lab in Zhengdian Industrial Park.Search for the Origin
Civets and Markets
The search for the origin of the new virus commenced. Live animal markets were an early focus, largely based on anecdote:
Professor Zhong Nanshan had reported that one of the earliest SARS cases in Heyuan was a chef who had come into regular contact with several types of live caged animals used as exotic game food. Because of this, Guan Yi and B.J. Zheng, who led the effort to identify the animal host of the SARS virus, focused their attention on wild animals recently captured and marketed for culinary purposes.
Guan Yi’s joint study with Guangdong CDC found closely related coronaviruses in samples from four palm civet and one raccoon dog (note the samples were collected by the Guangdong CDC and given to HKU to sequence). These sequences were very similar to human SARS but all contained a 29 nucleotide insert in ORF8 and the study concluded they were phylogenetically distinct from the human virus. When a human case was found carrying the variant with the 29 nucleotide insert it appeared that civets at the market had infected a human. A cull of farmed civets was carried out on the recommendation of Zhong Nanshan.
China’s Ministry of Agriculture organized a separate collaboration to investigate the origins, comprising researchers from several Chinese institutions and Australia’s CSIRO, as civet farmers were devastated by the cull. They sampled nearby farms and found no evidence of widespread infection of civets, suggesting instead that Guan Yi’s civets may have been infected while at the market by another species. The resulting paper “Antibodies to SARS Coronavirus in Civets” includes as authors CSIRO’s Gary Crameri, Bryan Eaton and Linfa Wang, and Changchun Tu (then at Changchun Agricultural University).
Further studies by outside groups have since determined that civets or other carnivores were more likely infected by humans and not vice versa.
HKU Microbiology Expansion in Aftermath of SARS
In 2005, the PRC Ministry of Science and Technology established the first State Key Laboratories outside the mainland. One was the State Key Laboratory of Emerging Infectious Diseases (SKL-EID) at HKU. Though not yet a full professor, Guan Yi was named co-director as reward for his work tracing SARS origins. In 2011 China started direct funding his lab to the tune of $5 million/year allowing considerable expansion of the number of researchers employed there.
More recently Guan Yi and his lab have co-authored with Major-General Wu-Chun Cao and other researchers from AMMS, the March 2020 paper “Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins”, purporting to show that pangolins were an intermediate host in a zoonotic origin of SARS-CoV-2. An earlier study on pangolin origin potential had been led by Yang Ruifu with assistance from South China Agricultural University (this will be covered in more detail in a future post).
HKU Microbiology was also the workplace of defector Li Meng-Yan, who has claimed that the senior lab directors (including Malik Peiris) were acting under CCP orders, and that she was given the task of fabricating evidence that racoon dogs were responsible for the SARS-CoV-2 outbreak - which she refused to do.Focus Turns to Bats
An expanded joint Chinese and CSIRO team experimentally infected civets with human variants of SARS, and it became an open question whether civets had originally infected humans, or vice versa. It suggested at most civets may have been an intermediate, rather than reservoir species. The search for the origin turned to bats which, as an animal reservoir, were well known to the CSIRO scientists who had recently worked on Hendra virus.
In following years, viruses related to SARS (SL-CoVs) were found in bats by different teams in diverse locations in China. Most of the sequences have high homology (genetic similarity) with each other, even those collected geographically distantly. They are certainly closely related to SARS, some parts of the genome are nearly identical (including much of S2 of spike). But relative to them the spike gene of SARS has large insertions in the Receptor Binding Motif (RBM) which is crucial for determining host and tissue tropism. These inserts are needed to allow the virus to bind to ACE2 receptors in the human (or civet) respiratory system.
It wasn’t until 2013 that a team comprising researchers from Wuhan Institute of Virology (including Zhengli Shi), EcoHealth Aliance (including Peter Daszak), CSIRO (Linfa Wang and Gary Crameri), announced they had found, in a bat fecal swab taken in a cave in Yunnan, a virus with very similar RBM inserts as SARS (Ge et al, 2013). The sample is named Rs3367, and the (near identical) virus they isolated they called WIV1. This is the foundational piece of evidence on which our assumption that SARS1 comes from a bat-hosted natural reservoir relies.

A few months later another sequence (LyRa11) was published that was also purported to have very similar RBM features, purporting to confirm the bat origin of the virus. The lead author of this sequence and accompanying paper was Changchun Tu, who stated his institutional affiliation as AMMS on that paper (it’s not clear when he became a PLA officer).
CSIRO Involvement with WIV and AMMS
In May 2020 journalist Sharri Markson wrote an article exposing the long friendship (and scientific collaboration) between AMMS' Changchun Tu and now Director of CSIRO’s Australian Animal Health Laboratory (AAHL), Trevor Drew. Although the article makes no specific allegations, information has been clearly leaked by someone close for unclear reasons. In the past Zhengli Shi and Peng Zhou from WIV have both worked and trained at AAHL. Linfa Wang, while a CSIRO employee, sat on WIV’s Scientific Advisory Committee alongside AMMS representatives Changchun Tu and Wuchun Cao.
As the only western government institution of the parties involved in SARS origins, I had hoped CSIRO would have retained documents that might shed light on the process and deliberations CSIRO parties leading to the WIV1 "discovery". Unfortunately to date they have been un-cooperative in responding to FOIA requests, claiming to hold no relevant documents or correspondence.
Although Linfa Wang departed CSIRO some years ago, Gary Crameri continues to author papers with him under a CSIRO affiliation. Some of these relate to SARS-CoV-2 and are in support of a natural origin. Despite this, CSIRO denies that Crameri has been employed by them since 2016. They also deny holding his emails, despite US Right To Know having previously obtained some by FOIA.Later, the WIV/EcoHealth/Linfa Wang (now at Duke-NUS) group claimed the discovery of two new viruses (RsSHC014 and WIV16) from the same cave. Ralph Baric took an interest and was given an isolate of WIV1 by Zhengli Shi. He also independently synthesized RsShC014, and confirmed for both the potential for spillover given the very similar RBM to human SARS.
Early Rumours of a Bioweapon
From the earliest days of the SARS outbreak rumours circulated in China that the new disease was the result of biological weapons research, and AMMS was high on the suspects list.
A Russian scientist Sergey Kolesnikov described SARS as “a cocktail of mumps and measles which could never appear in nature, only in a lab”. This was cursorily fact-checked and dismissed by media - of course measles and mumps are unrelated viruses. But it’s likely this reference was to SARS’ ability to bind to both a protein receptor (like measles) and sugars/sialic acids (like mumps). This is indeed unusual. As with the Covid-19 pandemic, the media “fact-checking” sound bites with no understanding of the context seems to have also been a problem.
General Xu Dezhong: Biowarfare Booster or Whistleblower?
In April 2014 a paper was published in English in Chinese Medical Journal by PLA epidemiologists from Fourth Military Medical University Shaanxi. The lead author is General (or Professor) Xu Dezhong. This paper provides succinct evidence of a theory doggedly advanced by Xu, that SARS was of unnatural origin:
“Based on the most abnormal characteristics in the existing findings on the epidemic, we could now state with certainty that the natural history of SARS is very unusual in epidemiology compared to that of other human infectious diseases, and furthermore, the conclusions should be extraordinary compared with those in the studies on SARS conducted so far.”
“We propose a new explanation about the origin and evolution of SARS CoV. SARS CoV was unnaturally produced and transmitted. It was continuously subjected to the great pressure of inadaptation and returned back gradually to its ancestor’s state through the reverse evolution. According to the research, we put forward the possibility of a global spreading of a zoonosis caused by the pathogen of unnatural origin.”
This paper appears to be a serious scientific work and discusses mutations in critical RBM residues and the 29 nt ORF8 insert in variants over the course of the pandemic. Xu observes that a later human strain found in Guangzhou, looks more like the earlier strain found only in civets. But the strains in the interim had been more severe and the 29nt insert had been deleted. He asks “how did the ancestral strain re-appear” - reverse evolution? I have some trouble reconciling this with other literature that suggests further deletion mutations in ORF8. These are also curious, but perhaps suggest a different evolutionary path.
Xu also noted that no natural zoonotic reservoir had been found, that bat SL-CoVs found to date were not similar enough to have caused the outbreak. The timing of its publication is unfortunate. Despite being published months after WIV1, it contains no mention of it, referring to another bat virus Rp3 as the closest ancestor known at the time:
“It seems that the great grandfather of SARS CoV has been found but not the grandfather and the father. Thus, we define Rp3 as “the parental generation (PG) 3” of SARS CoV, leaving “PG 1 and 2” to be found”
Xu had tried to get his work published as early as November 2012, and not in obscure Chinese military journals, but the Lancet.

The Lancet eventually declined to publish the paper after a three month delay.

A few weeks before Xu’s submission a group including Institut Pasteur, Shanghai and Zhong Nanshan had a pre-print accepted for publication by PLOS ONE. This paper added to the argument that SARS originated in bats, not civets, and was based on epidemiology and serology, not a new bat virus discovery. Shortly after Xu’s submission that decision was mysteriously rescinded, the authors were baffled:

On 16th May 2013, Nature received Ge et.al’s “Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor” (though the WIV1 sequence wasn’t published until 8th July, 2013, and the paper appeared in Nature on 30th November, 2013).
Xu also tried to alert the WHO to his findings, but was likewise brushed off:
“Through the research in this paper, we formally propose to WHO: to organize an expert committee, in conjunction with relevant governments and departments, to complete the following two tasks:
1. Conduct investigations and studies on relevant populations and animal groups, confirm that SARS-CoV has disappeared in humans and nature, and make an official announcement to reassure the public and the governments of relevant countries
2. In a specialized laboratory, verify the experimental process of bat Bt SL-CoVRp3 or similar viruses through unnatural modification to produce SARS-CoV.”

Xu’s papers don’t refer to bioweapons or nominate a suspect, and are sober in tone. But a subsequent e-book collection of papers with Xu as editor-in chief, but including many other authors and editors (including Yang Ruifu) - “The Unnatural Origin of SARS and New Species of Man-Made Viruses as Genetic Bioweapons” has been cited as blaming the US for SARS as a bioweapons attack, and exhorting China’s military to expand research efforts in biowarfare. While some papers give a backgound to trends in bioweapons research, papers in later part are related to Xu’s theory that SARS is artificial. My view is that Xu’s interest is only in having an artificial origin investigated. He likely isn’t privy to the bioweapons secrets of AMMS, nor does he seem to be blaming the US, or promoting a bioweapons arms race. This may not be true for AMMS officers.
Even after the publication of Ge et al, Xu didn’t back down, launching a robust critique of their work, which unfortunately doesn’t seem to have been published anywhere on the western internet:

One of Xu’s outstanding criticisms after the Rs3367/WIV1 claimed discovery is that the 29nt insert region in their ORF8 is the same as the other bat SL-CoVs, but different to those in human and civet SARS variants (in fact the entire ORF8 gene of the bat CoVs has low homology to SARS).

This was answered in early 2016 by a team from CAS Institute of Pathogen Biology with the announced discovery of new bat SL-CoVs which conveniently had the SARS like ORF8. The explanation given was that although viruses with this feature were rare, recombination with other CoVs must be much more common than previously thought. Perhaps one of these rare bat viruses recombined with the civet virus in a Guangdong market?
“Guangdong Province is the primary region in China in which wildlife (including bats) is consumed…Furthermore, the observation that SL-CoVs from R. sinicus are prone to recombine with CoVs from other hosts may suggest that the wildlife markets in Guangdong may provide an ideal incubator for the genesis of SARS-CoVs. Moreover, human consumption of wildlife increases the possibility of human exposure to viruses carried by wildlife.”

More Doubts Over the Evidence for Zoonosis
I hadn’t personally questioned the provenance of these sequences, and only recently read Xu’s work, but serendipitously I ran a comparison between them using SNAP. I was looking only to find an example comparing (I assumed) natural SL-CoVs. SNAP helps visualize differences between sequences after they’ve been aligned in another tool. It draws three lines representing cumulative mutations between the sequences. A green line shows synonymous mutations. These don’t change the protein encoded so make little difference to the virus’ evolutionary competitiveness. Whether such mutations survive and replicate is more or less random. So we expect them to be scattered roughly evenly through the gene and the line to be approximately straight.

Non-synonymous mutations are shown by a red line. These change the protein encoded so determine the evolutionary competitiveness of the mutant virus. Most changes are deleterious, so mutations rarely survive and replicate. But because the virus is engaged in a battle to evade the host immune system, changes in the immune exposed regions are more often beneficial. So the line is steeper in these parts (mostly in the first half of the spike - the NTD and RBD).
The third line (blue) shows inserts and deletions (indels). This type of mutation is quite rare. Many comparisons of related sequences show none, the line is usually flat, or nearly so.
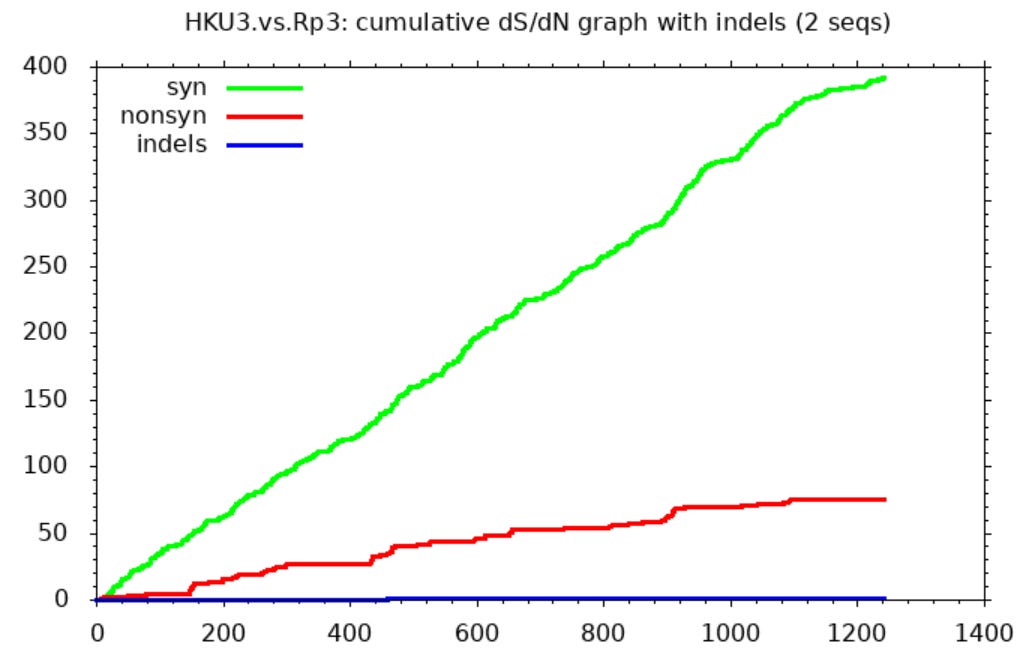
The SNAP sequence comparisons of Rs3367 (WIV1), RsSHC014 and WIV16 (and some other sequences published by this group) bear no resemblance to expectations for natural sequence pairs. The green line is flat for much of the gene indicating nearly identical sequences with one region highly variable between each pair (in this case the RBD between aas ~420-480). Outside this region there are just 2 non-synonymous, and one synonymous mutation in the rest of the spike. One way to attain this would be to take the spike RBD from a different virus (or a synthetic construct), and spliced it into a common backbone virus.
Interestingly Zhengli Shi and Linfa Wang had conducted a very similar experiment to this in 2007 replacing an almost identical RBD region into an SL-CoV backbone:
“In addition to full-length S of SL-CoV and SARS-CoV, a series of S chimeras was constructed by inserting different sequences of the SARS-CoV S into the SL-CoV S backbone…a minimal insert region (amino acids 310 to 518) was found to be sufficient to convert the SL-CoV S from non-ACE2 binding to human ACE2 binding, indicating that the SL-CoV S is largely compatible with SARS-CoV S protein both in structure and in function.”
Coincidence that they could find a natural recombinant exactly like this in a fecal sample taken in a bat cave?
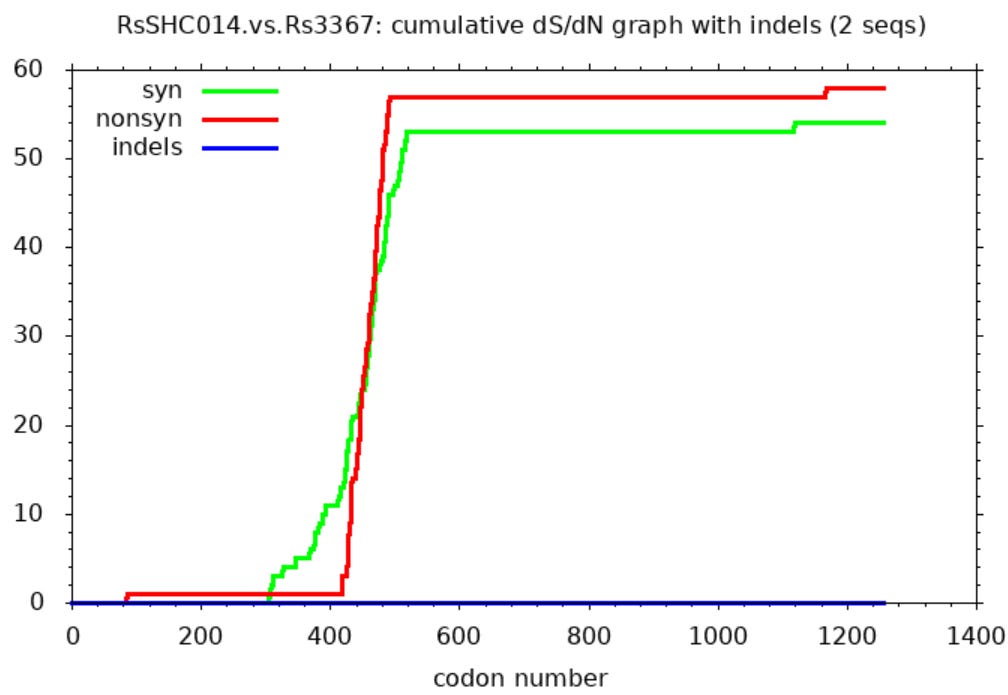
Comparing WIV1 with later discovery WIV16 the SNAP diagram looks similarly weird. This looks like someone has taken the NTD of a different virus and attached it to a common backbone spike. After the NTD there are just two non-synonymous mutations. There is also a curious region where non-synonymous mutations stop, but synonymous continue.

The intended, and only plausible natural explanation is that these are the result of recombination between different viruses. Although recombination is an accepted phenomenon, and this possibility shouldn’t be lightly dismissed, it does invite questions:
Recombination must have happened very recently as evidenced by the lack of synonymous mutations outside the variable regions.
The breakpoints occur in the same places someone interested in artificially engineering recombinants to develop specific tropism might choose.
There are areas of overlap where synonymous (green) mutations occur, but no non-synonymous. This suggests selective pressures apply within an unusually precise region.
The original viruses seem quite dissimilar, the lines are steep in the variable region. Recombination is usually thought to occur only between genetically similar species.
This suggests the possibility that one or more of WIV1, WIV16, RsSHC014 aren’t natural. If so, that undermines the assumption that SARS has a natural origin. The sequences (LyRa11, LyRa3) provided later by Changchun Tu and AMMS don’t suffer the same issues, but they are, after all, from an institution suspected of bioweapons development (and I believe much more technically capable than WIV).
The New (Pseudo?) Science of Recombination
Although recombination is a long-established phenomenon it might easily be abused in a motivated attempt to disprove that certain viruses aren't engineered. Previously scientists were careful to limit their claims to particular viral species, to study recombination mechanisms, analyze frequency, breakpoints, conditions which might favour this form of evolution. Recently recombination has become a catch-all for any genetic mutation that otherwise is difficult to explain. Claims are often self-referential or rely on precedents that themselves may be unreliable (e.g. the curious "natural evolution" of SARS-CoV-2 is often validated by reference to the "natural evolution" of SARS).
It isn't sufficient for scientists to claim "we know coronaviruses recombine frequently", without studies - preferably under controlled laboratory conditions - that show how frequently, at what positions, under what circumstances. And if a likely mechanism is shown to exist, the resultant recombinant virus must still be viable and competitive in the host environment in which it emerged. Viruses that evolved in the intestines of a bat are still unlikely to be well adapted for the human respiratory system, recombinant or not.
Unfortunately we must now also doubt the provenance of the data. Most sequences related to SARS like coronaviruses come from Chinese government sources - WIV, AMMS, CDC, CAS and others. Sometimes they come the imprimatur of western collaborators e.g. Edward Holmes, Linfa Wang, Guan Yi, EcoHealth, CSIRO. But does this guarantee samples or sequence data haven't been tampered with or fabricated, and genuinely reflect what was collected? There are no procedures in place that can assure this. Fabricating data is easy. Scientists must start to treat it with skepticism.
Unusual Molecular Features of SARS Spike Gene
Receptor Binding Motif
SARS’ RBM is compared below to a consensus sequence of bat SL-CoVs- excluding the dubious sequences with inserts WIV1, WIV16, LyRa11, RsShC014 - using the residues closest to those in the SARS sequence (Tor 2 variant is depicted).
Understanding Receptor Binding
The binding of two proteins such as receptors is sometimes illustrated using a "lock and key" metaphor, or perhaps an interlocking shape puzzle. But this is a bit misleading. Proteins are somewhat flexible, somewhat rigid, at a macroscopic scale easily understood as being a bit like meat. At a microscopic scale different amino acids vary enormously in properties like size, flexibility, the types and strength of their attraction to other amino acids, and affinity to water (hydrophobicity). And binding happens in 3D space so surface topology, the way each sequence is folded is also important. It's not sufficient to have pairs of amino acids attracted to each other, the spacing and alignment between them must also be conducive to bonding. It's rarely a case of all or nothing, binding affinity is expressed by relative strength measures.
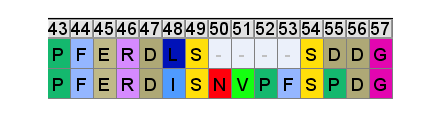
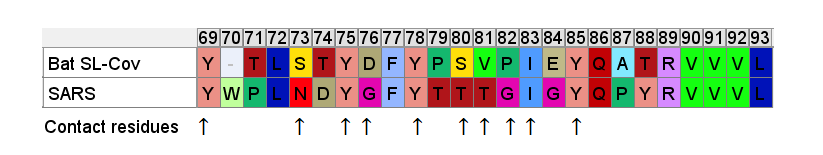
So even if we accept that an improbable insert mutation may have happened in a bat virus, it's hard to believe this would lead to a receptor binding domain that had high affinity for receptors common in the human respiratory system. Most likely in the very different host environment of a bat's guts, the mutant with such an insert would be discarded as at best useless, more likely deleterious.Zooming in, one interesting feature of the RBM of the bat SL-CoVs is a sequence of 3 consecutive Tyrosines (YYY). Tyrosine has properties that make it particularly useful in receptor binding. SARS also contain these 3 residues, but in SARS they are interspersed with others. There’s also an insert (ATS ) just upstream, and a fourth Y from a double substitution SH→YL
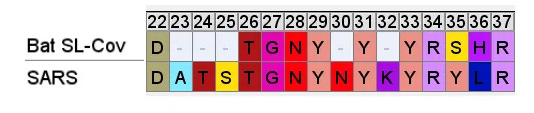
It's difficult for this to evolve in nature as there are 3 indels and 2 substitutions in the space of 10 residues. Yet there is clear homology, it’s unlikely the 3 Ys evolved independently. This is a critical region for binding. There are a handful of contact residues in the RBM experimentally known to directly bond with corresponding residues in ACE2, 3 are in this region. The increased spacing provided by the inserts and extra Y here are important for ACE2 binding strength. While difficult to understand how natural evolution would arrive at this, one mutation at a time, it is just the kind of thing an engineer might look to tweak to enhance binding affinity
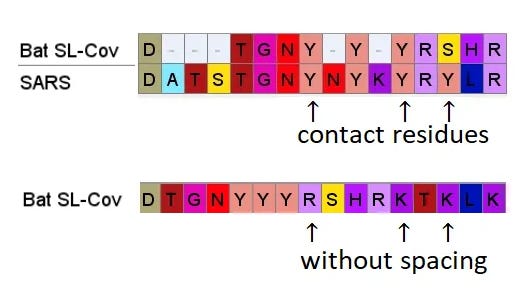
A few residues upstream is a possible inspiration for an engineer’s choice of inserted residues, another occurrence of the sequence YNYK. Coincidence? Perhaps. But similar copy/paste recurrences of short peptides are a characteristic feature of SARS spike gene.
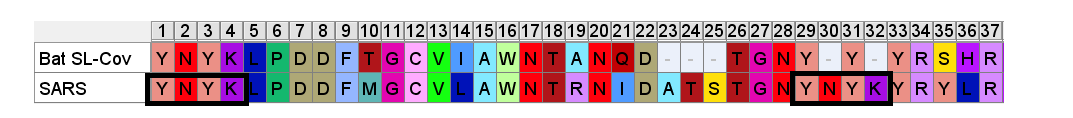
Another example of this sort of repeats: the next insert downstream includes a tripeptide SNV. SNV occurs 4 times in the first 460 amino acids of SARS, but just once in each of the bat SL-CoVs (in WIV1 there is even a 5th occurrence).

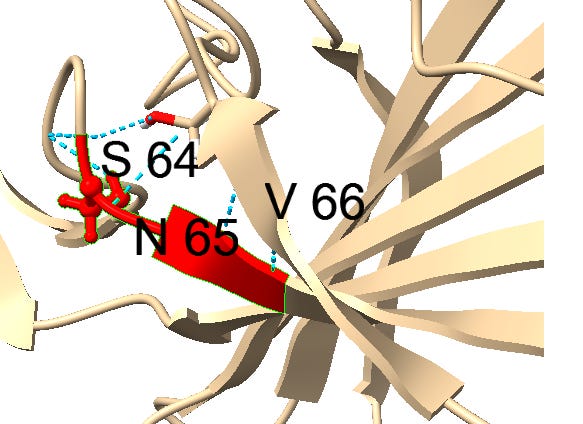
Though it’s not clear what the function of this motif is, it seems to have a similar structural context in most instances, occurring between ß-strands. It may have a role stabilizing turns with hydrogen bonds. Could there be a natural evolutionary explanation for this? A random 3 amino acid sequences occurs 1 in 8000 residues, so 4 occurrences in less than 500 residues is extremely unlikely if random. A template switching or RNA slippage mechanism is a possibility, but why would they appear in similar structural contexts?
Downstream of the SNV peptide is a loop insert featuring several Proline (P) residues. Proline has several features that may make it of interest to an engineer. It can provide unique structural rigidity. It can also have a binding role in itself, bringing together proteins rapidly so that other stronger bonds (such as between charged residues) can be formed. This structure may serve both roles, it also contains a pair of cysteine residues which bind each other with disulfide bonds providing additional structural support.

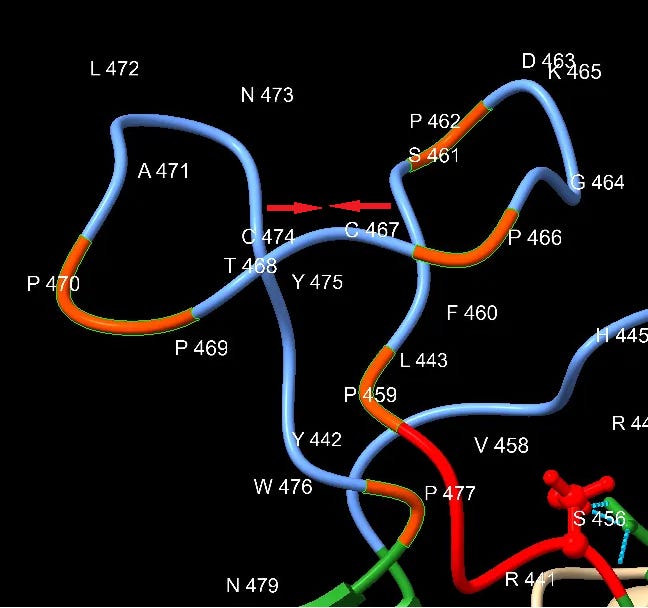
This appears quite a contrived structure. Is it possible this structure evolved in an ancestral virus, and has been deleted in the bat SL-CoVs? This should make us wonder why SARS hasn’t appeared in local populations (or elsewhere in the world), and why bat SL-CoVs with these inserts have been so difficult to find (and continue to be, outside China).
In 2010 a mostly German team led by Christian Drosten announced a surprising new discovery. A bat virus found in Bulgaria (BM48-31) had an RBM which, although different at the 5’ end and lacking the previous insert, had a similar insert loop in this region, including the cysteine pair. This, they claimed, gave it a closer RBM to SARS than any virus yet found in China, despite the sequence being quite different in many other regards (such as having no ORF8). This hints that an ancestor to the Chinese bat SL-CoVs may have had this loop and was subsequently deleted.

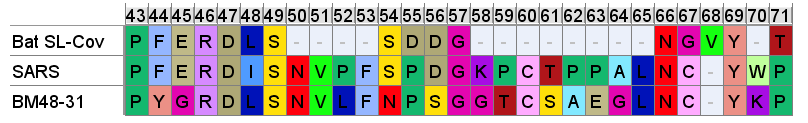
The timing of this discovery is interesting coming between WIVs work making synthetic RBM clones, in 2008, and their purported discovery of Rs3367 (in 2011). Also interesting is the presence of Hongkui Deng of Peking University as an author on both the Drosten paper, and the WIV cloning paper. This is another piece of research that isn’t quite clear of the WIV or Chinese state’s orbit. Before and since the discovery of BM48-31 several other SARS related viruses have been discovered in Europe, by authentically independent teams, but none have claimed to find similar RBM features.
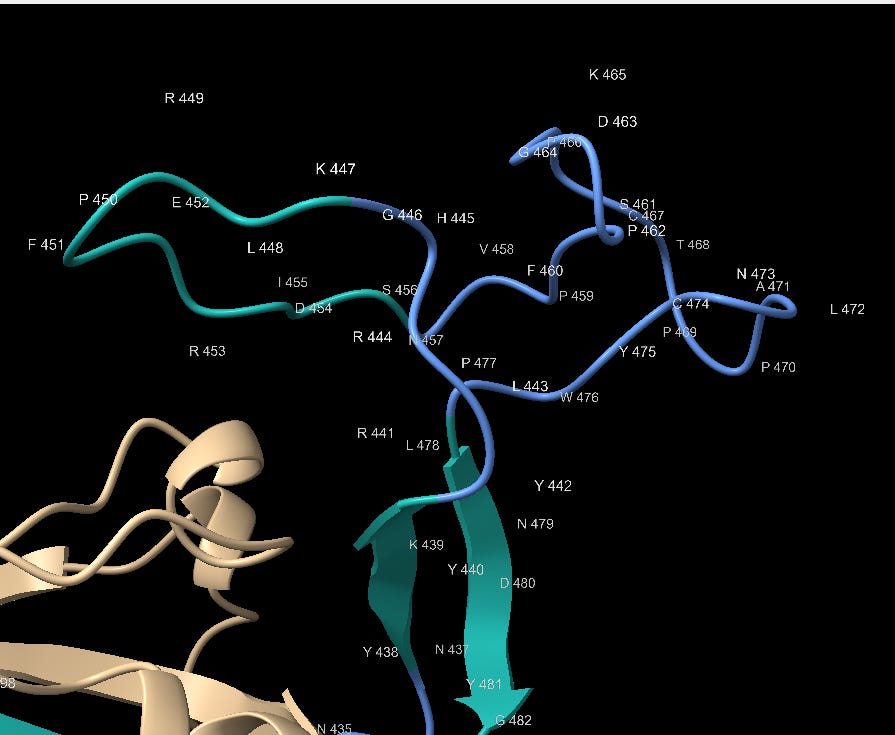
The 3’ end of the RBM is also interesting, having highest concentration of contact residues. Most of the Tyrosine (Y) residues are conserved with bat SL-CoVs but other residues are less so. SARS contains 3 Glycine (G) residues not present in the bat SL-CoVs. Glycine is a small amino acid, also providing conformational flexibility and often used in genetic engineering for these attributes. Here it may allow Tyrosine, other polar and non-polar residues flexibility to orient to the corresponding binding site.
An Alien Sequence in the N-Terminal Domain?
In the NTD (the “start” of the spike sequence) there are a number of surface exposed loops that are somewhat more variable between sequences. There is no homology between SARS and SARS-like bat viruses in this region, SARS contains a large deletion and a completely different sequence following the SNV (see above).

BLASTing the sequence GFHTINHT resulted in a very close match to human adenovirus D, more specifically type 37, a virus with no relation to coronaviruses. What could this be about? Adenoviruses can have very different symptoms and tissue tropism depending on some quite minor changes to the sequence. Type 37 is known to cause epidemic conjunctivitis and pharyngoconjunctival fever. It’s tissue tropism is broad, not only infecting the conjunctiva and cornea, but pharyngeal, intestinal, cervical and urethral membranes. This had been established in 2000 as due to its ability to bind sialic acids. A paper published in early 2002 a few months before SARS emerged, identified a specific gene as responsible for this.
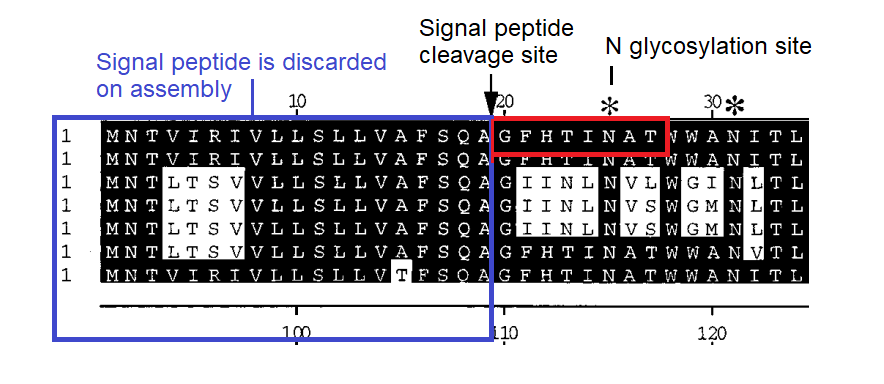
A diagram in the paper indicated the predicted signal peptide cleavage site showed that GFHTINAT marks the true start of the NTD of the protein, and that adenovirus types that contained this caused the severe conjunctivitis, while others without it do not. Note there is one amino acid different A→H but the N-glycosylation site surrounding it is conserved. See the box below for a discussion on the statistical significance of short sequences. A matching 8 amino acid sequence to an unrelated human virus would be a very improbable occurrence. Although this is not quite that, the position of the source sequence, the timing and topic of the Adenovirus publication, and the conserved glycosylation site should also be considered.
Assessing Probability of Short Insert Sequences Arising "At Random"
It is not easy to calculate how likely it is a short sequence may have arisen by natural evolutionary process in the spike gene. There are several considerations:
•It is simple enough to calculate the number of permutations of short sequences of amino acids. There are 20 amino acids so possible permutations in a sequence of 2 is 400, 3 is 8000, 4 is 160000, 5 is 3.2 million, 6 is 64 million, 7 is 1.28 billion, 8 is 25.6 billion. But this is only useful as a guide.
•The size of the dataset we are searching also matters. Genbank currently contains 10 trillion base pairs of data. It is highly likely that any short sequence will match something in a giant database. But little of this data is relevant to our search e.g. humans have genomes a million times larger than viruses and comparing to their genomes may not be relevant. If we restrict a search to the context we are interested in matches are much harder to come by. If we are only interested in the spike genes of human coronaviruses (there are only 7), the dataset we are searching is only ~8,750 amino acids in total. A match to a short sequence is far more significant in this context.
•Calculations are complicated further because evolution isn't random. certain amino acids may be favoured in some regions/genes/organisms over others. Convergent evolution is also a possibility.
•There are some helpful studies that can e.g. show statistically that insert mutations are rare, extremely so when larger than 4 amino acids. Even so these aren't quite relevant to our context.
In all assessing the likelihood of these mutations arising "randomly" is more art than science, and the context is always important. It's important to consider all relevant information, not treat each item of evidence separately.NTD Binding for a Dual Attack
While the paper doesn’t identify this peptide or any other region as important to the binding process there are many viruses that use the NTD as a binding site, and sometimes that is just the first few residues of the NTD (after the signal peptide is cleaved). A diagram from the paper “Receptor Recognition Mechanisms of Coronaviruses” shows various coronaviruses and the location of their known binding sites. I have updated to include more recent outbreaks:
Some viruses (e.g. HIV) are known to have a dual binding mechanism, attaching to different (co)-receptors in the NTD and RBD. An initial attachment via the NTD can trigger a conformational change before a stronger attachment is made via the main binding sequence in the CTD of S1. SARS and SARS-CoV-2 appear designed to do the same. Though the RBD in both appears well tuned to human ACE2, NTD binding regions might be less host-specific, recognizing sialic acids present also in animal hosts.
Comparing this NTD loop in SARS to corresponding sites in SARS-CoV-2 and some other more recent sequences is also interesting. There is interesting sequence homology between SARS-CoV-2 and a sialic acid binding region from unrelated virus MERS. There is also homology between this MERS region and ZC45 (a sequence published by PLA scientists from Nanjing Command in 2018). The SARS-CoV-2 version of the region looks like a mash-up of three different sequences. Not the sort of reshuffling we would expect to occur naturally. Convergent evolution is one possibility, however there are reasons to think ZC45 is also unnatural (evidence for this in a forthcoming post).

Could there be other engineered viruses?
In 1999 researchers led by Dutch scientist Peter Rottier switched the spike of Mouse Hepatitis Virus (MHV) for that of Feline Infectious Peritonitis Virus (FIPV) and found that it now infected cats, not mice. At the time there were only two known human coronaviruses. Within 6 years there were three new HCovs with signs of zoonotic ancestry: SARS, HKU1, NL63. The latter two haven’t caused severe epidemics, usually symptoms are mild, but can pose a serious health risk to people with weakened immune systems, infants and the elderly. Seroprevalence is high, most of us have unknowingly already had them, and likely experienced them as little more than a cold.
HKU1 emerged in the same region as SARS, was first identified in an elderly Hong Kong man who had returned from a trip to Shenzhen. It has several features that make it suspicious as a lab construct. NL63 was notable for also using the ACE2 receptor. I’ve written briefly about some of HKU1’s unusual genomic features.
Out of the Frying Pan, into the Fire
In 2017 WIV (with EcoHealth and Linfa Wang’s stamp of approval and funding) published “Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus” which claimed to finally solve the mysteries of SARS origin by finding what seems to be “Magical Cave of Recombination” containing “all the building blocks” needed to assemble SARS and potentially other novel pathogens:
“As a whole, our findings from a 5-year longitudinal study conclusively demonstrate that all building blocks of the pandemic SARS-CoV genome are present in bat SARSr-CoVs from a single location in Yunnan. The data show that frequent recombination events have happened among those SARSr-CoVs in the same cave. While we cannot rule out the possibility that similar gene pools of SARSr-CoVs exist elsewhere, we have provided sufficient evidence to conclude that SARS-CoV most likely originated from horseshoe bats via recombination events among existing SARSr-CoVs. In addition, we have also revealed that various SARSr-CoVs capable of using human ACE2 are still circulating among bats in this region. Thus, the risk of spillover into people and emergence of a disease similar to SARS is possible.”
And this prediction proved to be correct. By duping western scientists into accepting “recombination” as an explanation for all genetic peculiarities without skepticism, and accepting any sequence published without questioning, they have enabled pathogens to be manipulated in bioweapons labs and even deliberately released without fear of accountability. And so the seeds for the Covid-19 pandemic were sown.
Thoroughly enjoyable read. It's refreshing to find so much detail presented in such a clear and accessible form.
Once you see these similarities laid out, it's striking how such a comparative analysis hasn't been attempted before now. I hope this stimulates lots of discussion and even further analysis.
Two of the characters you mention have not gained as much attention as the likes of Fauci, Daszak, Shi or even Baric - I'm referring to Lin-Fa Wang and Garry Crameri - their role in the SARS-COV-2 drama is worthy of scrutiny imho.
Congrats on your first post, welcome to substack and I hope we'll have a chance to read more from you.
The following code downloads FASTA files for nucleotide and amino acid sequences of SARS-like viruses, it aligns the spike protein sequences, and it sorts the sequence by their number of mismatches to Tor2 in the region which features the DATSTGNYNYKYRYLR sequence in Tor2:
brew install mafft seqkit brewsci/bio/snp-dists xmlstarlet
curl -Lso sarslike.fa 'https://drive.google.com/uc?export=download&id=1j-YFiMYG4DkVKSget2fYW-gaJDy6NCkW' # 335 aligned sequences of SARS-like viruses from GenBank
curl 'https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&rettype=fasta_cds_aa&id='$(seqkit seq -ni sarslike.fa|paste -sd, -)>sarslike.aa
seqkit grep -nrp spike\|surface sarslike.aa|mafft ->spike.aln
snp-dists sarslike.fa>sarslike.dist
xml fo -D sarslike.xml|xml sel -t -m //GBSeq -v GBSeq_accession-version -o $'\t' -v GBSeq_definition -o $'\t' -v GBSeq_create-date -o $'\t' -v './/GBQualifier[GBQualifier_name="collection_date"]/GBQualifier_value' -o $'\t' -v '(.//GBAuthor)[1]' -o ... -v '(.//GBAuthor)[last()]' -o $'\t' -v '(.//GBReference_title[text()!="Direct Submission"])[last()]' -o $'\n'>sarslike.tsv
tab(){ awk '{if(NF>m)m=NF;for(i=1;i<=NF;i++){a[NR][i]=$i;l=length($i);if(l>b[i])b[i]=l}}END{for(h in a){for(i=1;i<=m;i++)printf(i==m?"%s\n":"%-"(b[i]+n)"s",a[h][i])}}' "${1+FS=$1}" "n=${2-1}";} # `tab \\t` is like `column -ts$'\t'` but it doesn't get thrown off by empty fields
x=NC_004718.3;seqkit subseq -r490:506 spike.aln|seqkit fx2tab|sed $'s/_prot_[^\t]*//;s/lcl|//'|gawk '{l=length($2);for(i=1;i<=l;i++)a[$1][i]=substr($2,i,1);b[$1]=$2}END{for(i in a){d=0;for(j=1;j<=l;j++)if(a[targ][j]!=a[i][j])d++;print i"\t"b[i]"\t"d}}' targ=$x|awk 'NR==FNR{a[$1]=$2;next}{print$3,$2,a[$1],$1}' {,O}FS=\\t <(seqkit seq -n sarslike.fa|sed $'s/ /\t/;s/, complete genome//') -|sort -n|awk -F\\t 'NR==FNR{a[$1]=$2;next}{print a[$4]"\t"$0}' <(awk -F\\t 'NR==1{for(i=2;i<=NF;i++)if($i==x)break;next}{print$1 FS$i}' x=$x sarslike.dist) -|sort -n|awk 'NR==FNR{a[$1]=$3 FS$4 FS$5;next}{print$0"\t"a[$NF]}' {,O}FS=\\t sarslike.tsv -|tab \\t
I posted the output of the shell commands here: https://pastebin.com/raw/GDm9PNqD.
Eight bat SARS viruses featured the sequence DATSTGNHNYKYRYLRH which has only one mismatch: BtRs-BetaCoV/YN2018B, Rs9401, Rs7327, YN2016C, YN2016D, YN2016E, YN2016A, YN2016B. They all have between 1254 and 1283 nucleotide changes from Tor2. WIV1 has about a hundred fewer nucleotide changes from Tor2 (1150) but it has two mismatches (DATQTGNYNYKYRSLRH). The only genome with three mismatches is "Rhinolophus affinis coronavirus isolate LYRa11" (DATSSGNFNYKYRSLRH), where the number of mismatches is pretty low considering that the whole genome has 2672 nucleotide changes from Tor2. The LYRa11 sequence was published in 2014 as part of a paper titled "Identification of Diverse Alphacoronaviruses and Genomic Characterization of a Novel Severe Acute Respiratory Syndrome-Like Coronavirus from Bats in China".
The Y?Y?Y pattern of three Y residues interspaced by single other residues is also featured in Wuhan-Hu-1: DSKVGGNYNYLYRLFRK. The region is identical in BANAL-52, BANAL-236, and BANAL-103. But in RaTG13 the first four residues DAKE instead of DSKV. And ZC45 has deletions in the middle of the sequence: "DV---GN--YFYRSHRS".